Barbed and ready
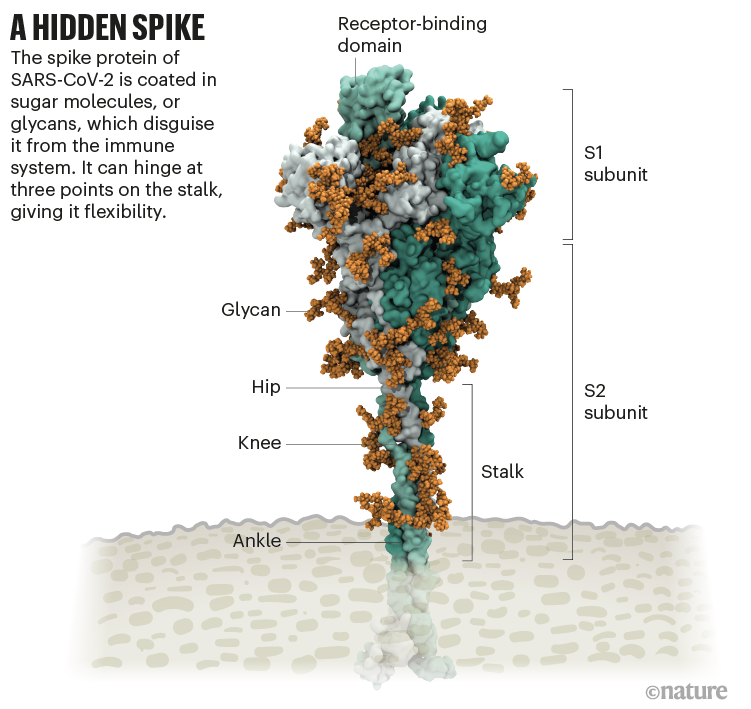
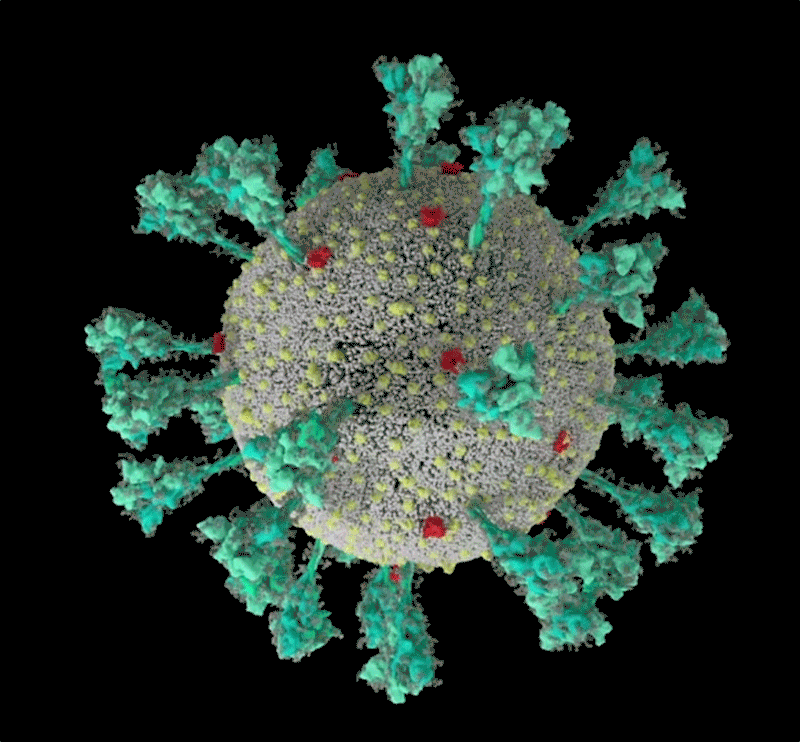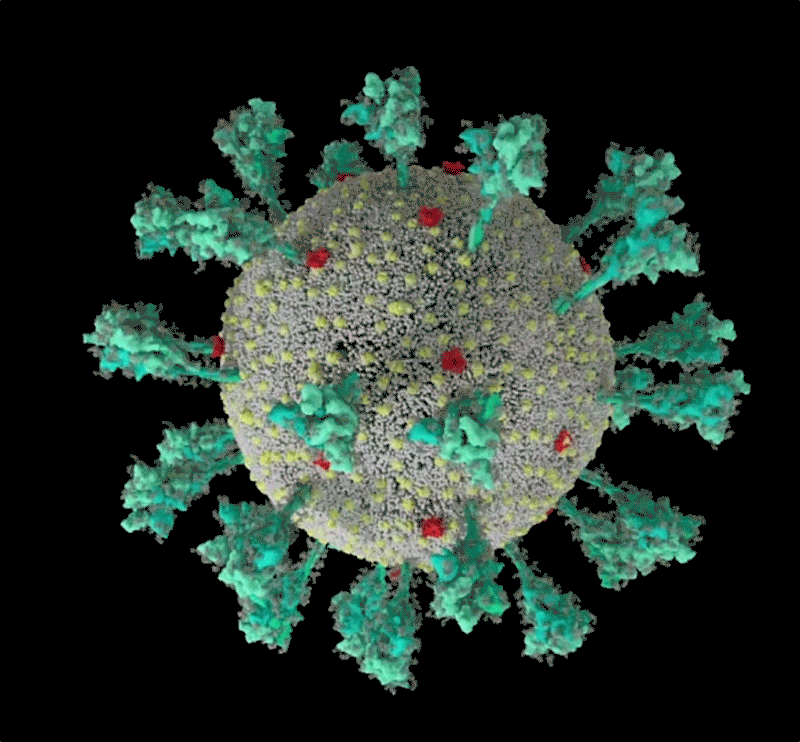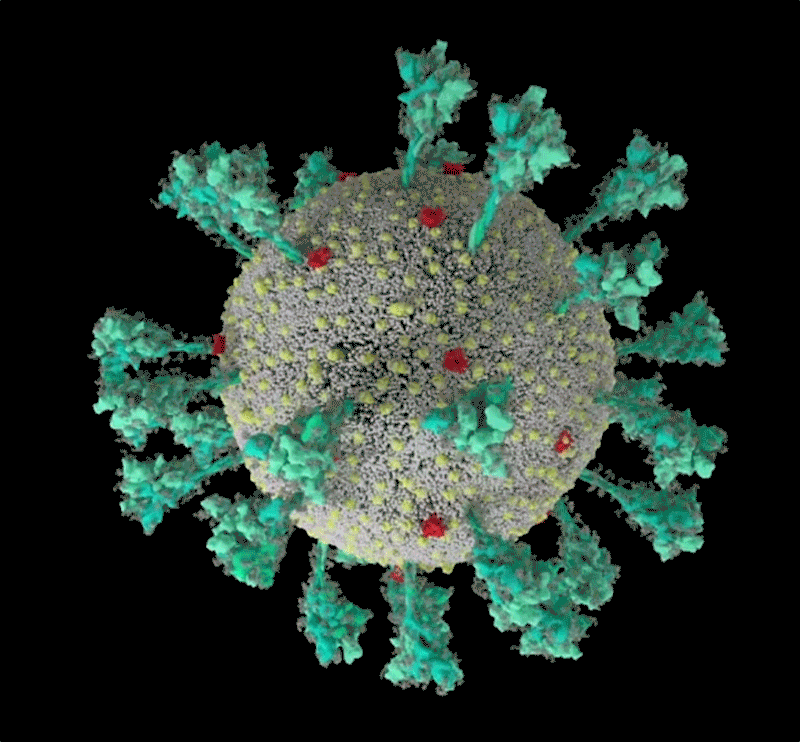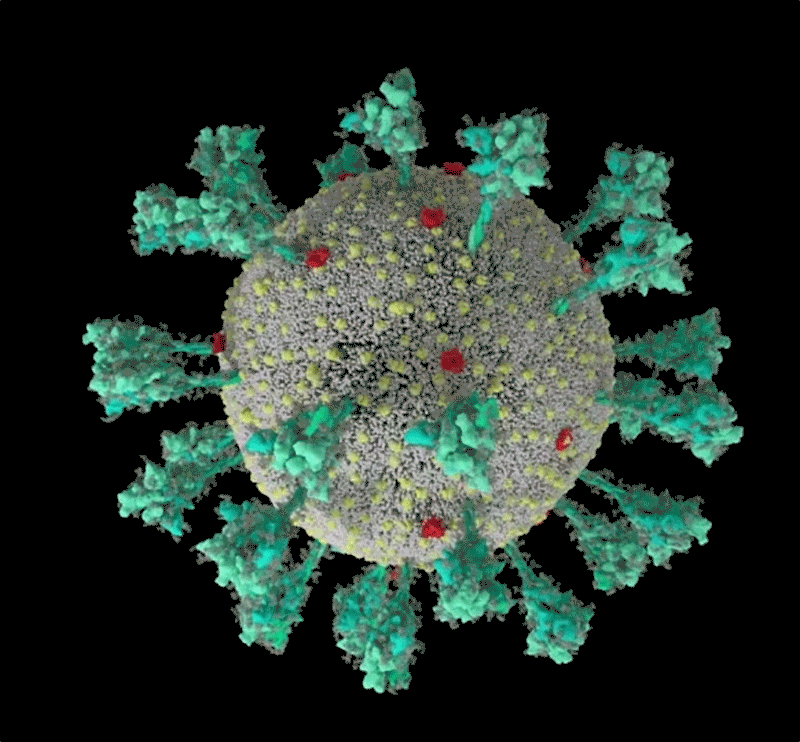
It starts with the spikes. Each SARS-CoV-2 virion (virus particle) has an outer surface peppered with 24–40 haphazardly arranged spike proteins that are its key to fusing with human cells2. For other types of virus, such as influenza, external fusion proteins are relatively rigid. SARS-CoV-2 spikes, however, are wildly flexible and hinge at three points, according to work published in August 2020 by biochemist Martin Beck at the Max Planck Institute of Biophysics in Frankfurt, Germany, and his colleagues3.
That allows the spikes to flop around, sway and rotate, which could make it easier for them to scan the cell surface and for multiple spikes to bind to a human cell. There are no similar experimental data for other coronaviruses, but because spike-protein sequences are highly evolutionarily conserved, it is fair to assume the trait is shared, says Beck.
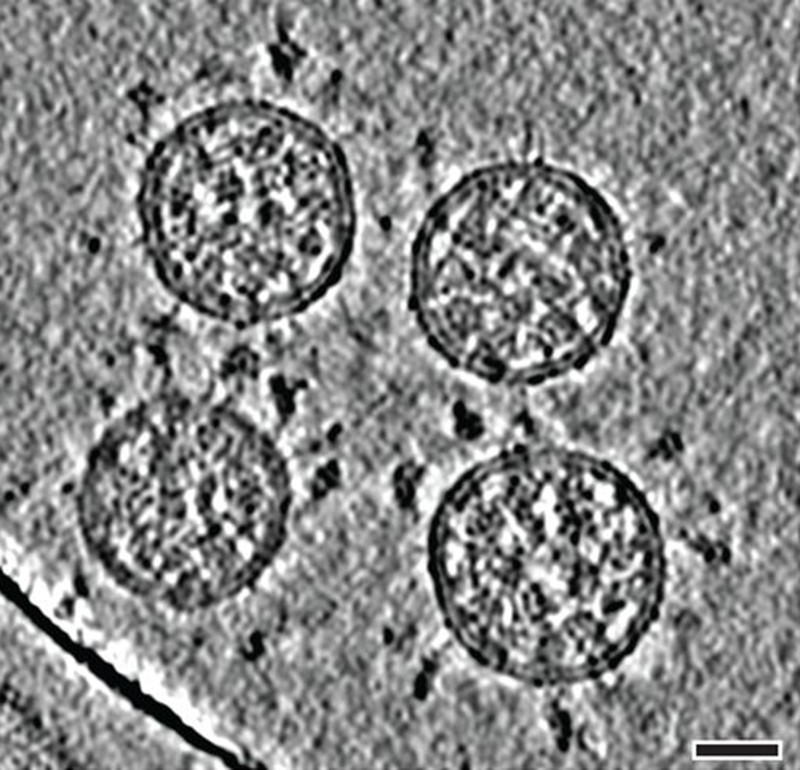
Cryo-electron tomography images of SARS-CoV-2 virions. (Scale bar: 30 nanometres.)Credit: B. Turoňová et al./Science
Early in the pandemic, researchers confirmed that the RBDs of SARS-CoV-2 spike proteins attach to a familiar protein called the ACE2 receptor, which adorns the outside of most human throat and lung cells. This receptor is also the docking point for SARS-CoV, the virus that causes severe acute respiratory syndrome (SARS). But compared with SARS-CoV, SARS-CoV-2 binds to ACE2 an estimated 2–4 times more strongly4, because several changes in the RBD stabilize its virus-binding hotspots5.
Worrying variants of SARS-CoV-2 tend to have mutations in the S1 subunit of the spike protein, which hosts the RBDs and is responsible for binding to the ACE2 receptor. (A second spike subunit, S2, prompts viral fusion with the host cell’s membrane.)
The Alpha variant, for example, includes ten changes in the spike-protein sequence, which result in RBDs being more likely to stay in the ‘up’ position6. “It is helping the virus along by making it easier to enter into cells,” says Priyamvada Acharya, a structural biologist at the Duke Human Vaccine Institute in Durham, North Carolina, who is studying the spike mutations.
The Delta variant, which is now spreading around the world, hosts multiple mutations in the S1 subunit, including three in the RBD that seem to improve the RBD’s ability to bind to ACE2 and evade the immune system7.
Restricted entry
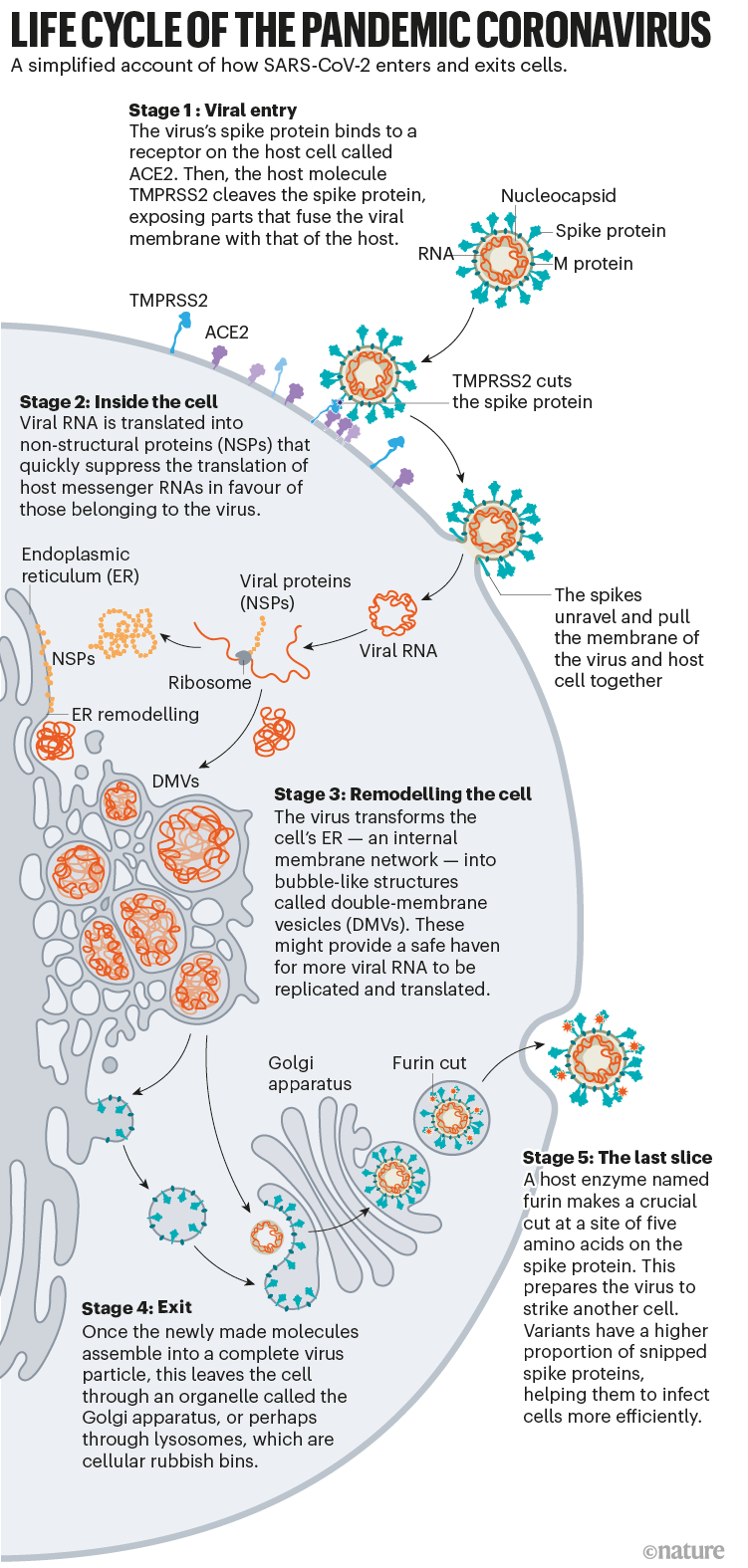
Once the viral spikes bind to ACE2, other proteins on the host cell’s surface initiate a process that leads to the merging of viral and cell membranes (see ‘Viral entry up close’).
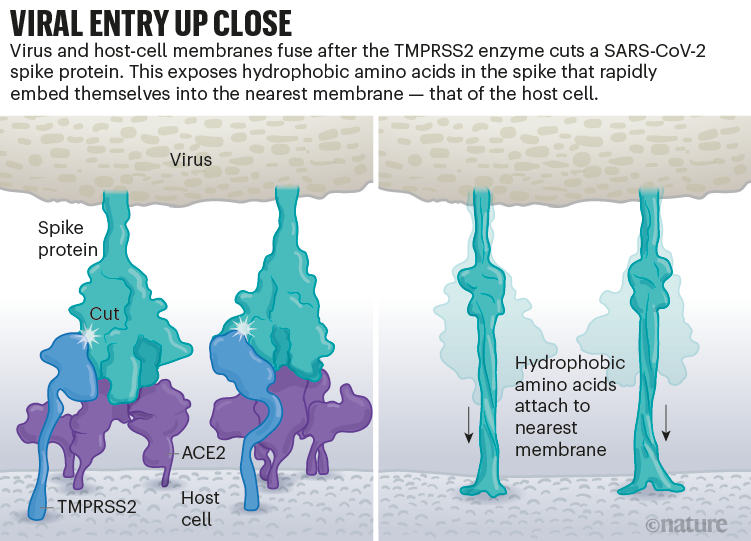
Source: Janet Iwasa, Univ. Utah; Graphic: Nik Spencer/Nature
The virus that causes SARS, SARS-CoV, uses either of two host protease enzymes to break in: TMPRSS2 (pronounced ‘tempress two’) or cathepsin L. TMPRSS2 is the faster route in, but SARS-CoV often enters instead through an endosome — a lipid-surrounded bubble — which relies on cathepsin L. When virions enter cells by this route, however, antiviral proteins can trap them.
SARS-CoV-2 differs from SARS-CoV because it efficiently uses TMPRSS2, an enzyme found in high amounts on the outside of respiratory cells. First, TMPRSS2 cuts a site on the spike’s S2 subunit8. That cut exposes a run of hydrophobic amino acids that rapidly buries itself in the closest membrane — that of the host cell. Next, the extended spike folds back onto itself, like a zipper, forcing the viral and cell membranes to fuse.
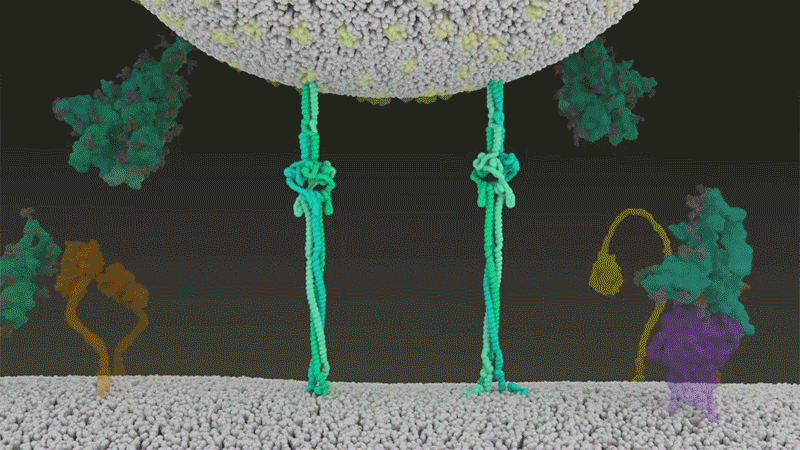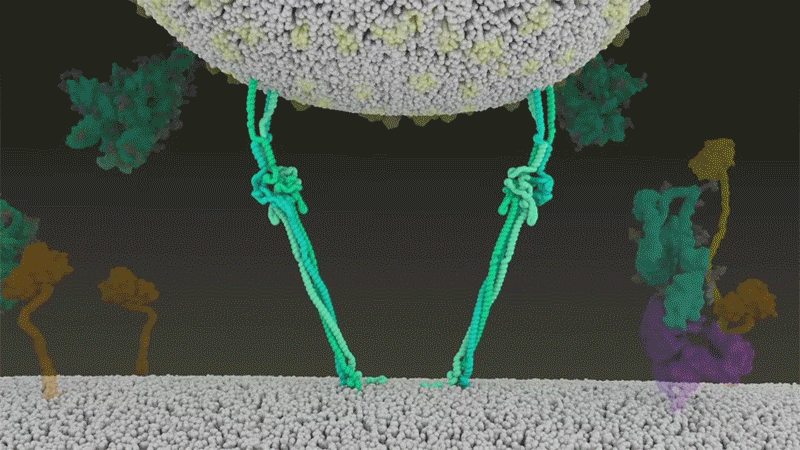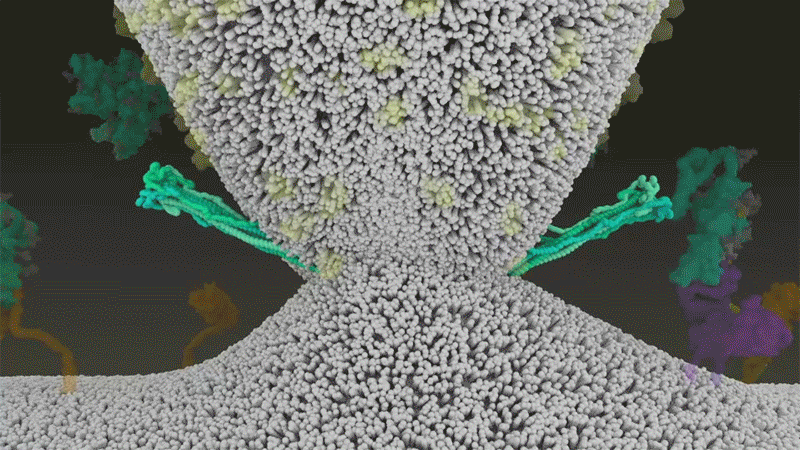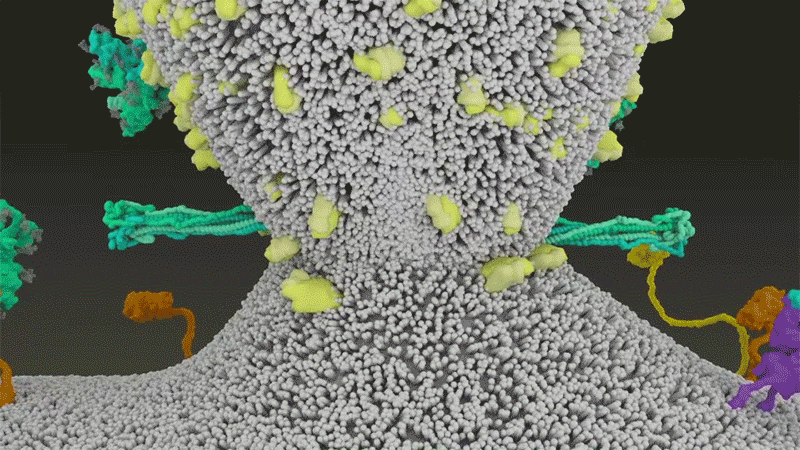
An animation of the way SARS-CoV-2 fuses with cells.Credit: Janet Iwasa, University of Utah
The virus then ejects its genome directly into the cell. By invading in this spring-loaded manner, SARS-CoV-2 infects faster than SARS-CoV and avoids being trapped in endosomes, according to work published in April by Barclay and her colleagues at Imperial College London9.
The virus’s speedy entry using TMPRSS2 explains why the malaria drug chloroquine didn’t work in clinical trials as a COVID-19 treatment, despite early promising studies in the lab10. Those turned out to have used cells that rely exclusively on cathepsins for endosomal entry. “When the virus transmits and replicates in the human airway, it doesn’t use endosomes, so chloroquine, which is an endosomal disrupting drug, is not effective in real life,” says Barclay.
The discovery also points to protease inhibitors as a promising therapeutic option to prevent a virus from using TMPRSS2, cathepsin L or other proteases to enter host cells. One TMPRSS2 inhibitor, camostat mesylate, which is approved in Japan to treat pancreatitis, blocked viral entry into lung cells8, but the drug did not improve patients’ outcomes in an initial clinical trial11.
“From my perspective, we should have such protease inhibitors as broad antivirals available to fight new disease outbreaks and prevent future pandemics at the very beginning,” says Stefan Pöhlmann, director of the Infection Biology Unit at the German Primate Center in Göttingen, who has led research on ACE2 binding and the TMPRSS2 pathway.