Overview
- On 26 November 2021, the World Health Organization ( W.H.O.) designated the
- variant B.1.1.529 a variant of concern (VOC), on the basis of advice from WHO’s
- Technical Advisory Group on Virus Evolution.
- The variant has been given the name Omicron.
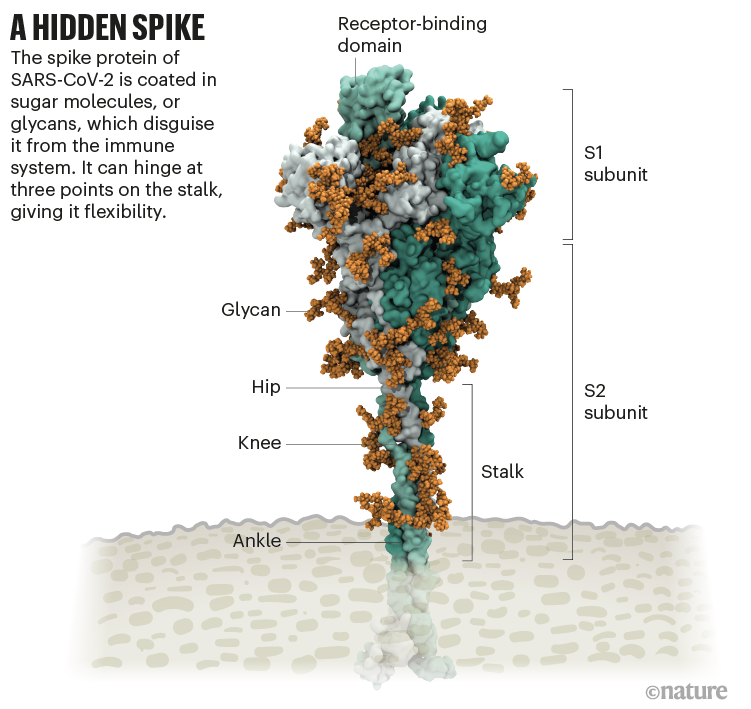
- Omicron variant is a highly divergent variant with a high number of mutations, including
- 26-32 in the spike protein, some of which are concerning and may be associated with
- immune escape potential and higher transmissibility.
- However, there are still considerable uncertainties.
- As of 9 December 2021, cases of human infections with this variant have been identified
- in 63 countries across all six WHO regions.
- Current understanding of the Omicron variant from recent data are likely to evolve as
- more data becomes available.
- The overall threat posed by Omicron largely depends on three key questions,
- including: (1) how transmissible the variant is;
- (2) how well vaccines and prior infection protect against infection, transmission,
- clinical disease and death; and (3) how virulent the variant is compared to other variants. Public health advice is based on current information and will be tailored as more evidence
- emerges around those key questions.
- Based on current limited evidence Omicron appears to have a growth advantage over
- Delta. It is spreading faster than the Delta variant in South Africa where Delta circulation
- was low, but also appears to spread more quickly than the Delta variant in other countries
- where the incidence of Delta is high, such as in the United Kingdom.
- Whether Omicron’s observed rapid growth rate in countries with high levels of population
- immunity is related to immune evasion, intrinsic increased transmissibility,
- or a combination of both remains uncertain. However, given the current available data,
- it is likely that Omicron will outpace the Delta variant where community transmission occurs.
- There are still limited data on the clinical severity of Omicron.
- While preliminary findings from South Africa suggest it may be less severe than Delta,
- and all cases reported in the EU/EEA to date have been mild or asymptomatic,
- it remains unclear to what extent Omicron may be inherently less virulent.
- More data are needed to understand the severity profile.
- There are limited available data, and no peer-reviewed evidence, on vaccine efficacy or
- effectiveness to date for Omicron. Preliminary evidence, and the considerably altered
- antigenic profile of the Omicron spike protein, suggests a reduction in vaccine efficacy
- against infection and transmission associated with Omicron.
- There is some preliminary evidence that the incidence of reinfection has increased
- in South Africa, which may be associated with humoral (antibody-mediated) immune
- evasion. In addition, preliminary evidence from a few studies of limited sample size
- have shown that sera obtained from vaccinated and previously infected individuals had
- lower neutralization activity (the size of the reduction ranges considerably) than with
- any other circulating VOCs of SARS-CoV-2 and the ancestral strain.
- The diagnostic accuracy of routinely used PCR and antigen-based rapid diagnostic test
- (Ag-RDT) assays does not appear to be influenced by Omicron.
- Most Omicron variant sequences reported include a deletion in the S gene, causing
- some S gene targeting PCR assays to appear negative.
- Although some publicly shared sequences lack this deletion, this remains a minority of
- currently available sequences, and S gene target failure (SGTF) can therefore be used
- as a useful proxy marker of Omicron, for surveillance purposes.
- However, confirmation should be obtained by sequencing, as this
- deletion can also be found in other VOCs (e.g., Alpha and subsets of Gamma and Delta).
- Therapeutic interventions for the management of patients with severe or critical
- COVID-19 associated with the Omicron variant that target host responses
- (such as corticosteroids, and interleukin 6 receptor blockers and prophylaxis with
- anticoagulation) are expected to remain effective.
- However, monoclonal antibodies will need to be tested individually, for their
- antigen binding and virus neutralization and these studies should be prioritized.
WHO TEAM
WHO Headquarters (HQ), WHO Health Emergencies Programme
EDITORS
World Health Emergencies Programme
NUMBER OF PAGES
2